در این مقاله، یک شرح تاریخی از توسعه رویکرد اوربیتال مولکولی، شامل نه تنها آلیاژهای Ti بلکه سوپرآلیاژهای پایه Ni ارائه خواهد شد. این شرح تاریخی تمام ویژگیهای نظریه طراحی آلیاژ ساخته شده بر اساس روش اوربیتال مولکولی را نشان می دهد. سپس پایداری فازی و مقاومت در برابر خوردگی آلیاژهای تیتانیوم در این روش توضیح داده خواهد شد، زیرا این خصوصیات برای طراحی آلیاژهای تیتانیوم برای کاربردهای بیوتکنولوژی مهم هستند. همچنین، با استفاده از نمونه ای از آلیاژهای Ti نوع β با استحکام بالا، یک روش مشخص برای طراحی آلیاژ نشان داده خواهد شد. سپس آلیاژهای تیتانیوم بیوتکنولوژی تولید شده توسط این روش ضمن استفاده از پیشرفت اخیر در این روش در طراحی آلیاژ بیوتکنولوژی، مرور خواهند شد.
تئوری طراحی آلیاژ
پارامترهای آلیاژسازی
خصوصیات فیزیکی و شیمیایی فلزات و آلیاژها با وضعیت الکترونیکی آنها ارتباط نزدیکی دارند. بیشتر آلیاژهای تجاری موجود از چندین جزء تشکیل شده اند. برخی آلیاژها از حدود 10 عنصر تشکیل شده اند. برای چنین آلیاژهای پیچیده، بسیار دشوار است که محاسبه دقیق حالت الکترونیکی به روشی معقول انجام شود. حتی اگر محاسبه به اتمام برسد، ممکن است برای طراحی آلیاژ کاربردی بی معنی باشد زیرا تعداد بی نهایت هایی از انوع و ترکیبات آلیاژی در آلیاژهای چند جزئی وجود دارد.
برای طراحی آلیاژ کاربردی، استفاده از پارامترهای آلیاژیی که ویژگی هر عنصر در یک فلز مادر را به خوبی نشان می دهد مناسب است. با وجود این، هیچ پارامتر آلیاژی با وجود سابقه طولانی متالورژی و علوم فلز یافت نشده است. بنابراین، بیشترین خصوصیات آلیاژ با استفاده از پارامترهای اختصاص داده شده به فلزات خالص محاسبه شده است (به عنوان مثال، شعاع اتمی، الکتروونگاتیوی، نسبت الکترون به اتم (e/a)) شد [4-5]. به عبارت دیگر، همواره مقدار یکسان پارامتر برای همان عنصر استفاده می شود حتی اگر سیستم آلیاژ متنوع باشد. از آنجا که هیچ اثر آلیاژی در پارامتر خالص دخیل نیست، پیش بینی خواص آلیاژ به ناچار در این روش ضعیف است. برای حل این مشکل، ابتدا باید پارامترهای آلیاژی را بصورت تئوری با استفاده از روش های اوربیتال مولکولی تعیین کنیم.
برای این منظور از دسته روش DV-Xα استفاده شده است. این روش اصل اول محاسبه اوربیتال مولکولی است. در مقایسه با روش محاسبه باند معمولی، این روش برای شبیه سازی ساختارهای الکترونیکی محلی در اطراف عناصر آلیاژی در فلز مناسب تر است. توضیحات بیشتر بیشتر در مورد دسته روش DV-Xα در جاهای دیگر آورده شده است [6-7].
آلیاژهای بر پایهی فلز انتقالی (به عنوان مثال آلیاژهای Fe و آلیاژهای Ti) در زندگی روزمره ما بسیار متداول هستند. برای طراحی آلیاژ، انتخاب پارامترهای آلیاژی متناسب با کاربرد آلیاژها بسیار مهم است. برای استفاده ساختاری و بیوتکنولوژی، برای طراحی آلیاژهای بر پایهی فلز انتقالی دو پارامتر آلیاژی ضروری است [8-10].
یکی از پارامترهای آلیاژی، ترتیب پیوند است (از این پس به آن Bo گفته می شود). همانطور که در شکل 1-3-1A نشان داده شده است، Bo میزان همپوشانی بین مدار اتمی ФΜ برای عنصر آلیاژی (M) و ФX برای فلز مادر (X) نشان می دهد. با افزایش جمعیت همپوشانی، استحکام باند کووالانسی بین اتمهای M و X افزایش می یابد. هنگامی که هر دو M و X فلزات انتقال هستند، پیوند کووالانسی d-d بین آنها غالب است. مقدار Bo بزرگتر به معنای پیوند شیمیایی قوی تر بین M و X است.
یکی دیگر از پارامترهای آلیاژی، سطح انرژی مدار d (از این پس به آن Md گفته می شود). همانطور که در شکل 1-3-1B نشان داده شده است، مدار d از اتمهای تنهای M و X ترکیب میشوند تا سطح پیوند در انرژی پایین تر و سطح بدون-پیوند در انرژی بالاتر تشکیل شود. هنگامی که سطح d یک اتم تنهای M بالاتر از یک اتم تنهای X باشد، انتقال بار از M به X انجام می شود تا باعث کاهش انرژی شود. در نتیجه، بار مؤثر برای M مثبت و برای X منفی می شود. از این طریق سطح انرژی جهت انتقال بار را کنترل میکند؛ از این رو این یک اندازه گیری برای الکترونگاتیوی است. عنصر با الکترونگاتیوی پایین تر از سطح انرژی مدار d (Md) بالاتری برخوردار است.
همچنین، خاطر نشان شده است که Md بسته به شعاع اتمی متفاوت است. هر اتم بزرگتر، شعاع متوسط مدار d بزرگتری دارد. در این حالت، با افزایش میانگین فاصله بین الکترونهای d و هسته در مرکز، نیروی جذبکنندهی کولمب که بین آنها کار می کند ضعیف می شود. در نتیجه با افزایش شعاع اتمی، سطح مدار d (Md) زیاد میشود. ذکر این نکته حائز اهمیت است که Md هم با شعاع اتمی و هم با الکترونگاتیوی مرتبط است، پارامترهای کلاسیکی که برای درمان مشکل حلالیت جامد آلیاژها مورد استفاده بوده اند [4-5].
هر دو Md و Bo پارامترهای آلیاژی جدیدی هستند که در ابتدا از محاسبه ساختار الکترونیکی تعیین می شدند. از آنجا که اینها پارامترهای الکترونیکی هستند، بزرگی آنها به ترتیب عناصر در جدول تناوبی تغییر می کند. با یادآوری اینکه اکثر خصوصیات فیزیکی و شیمیایی به دنبال جدول تناوبی تغییر می کنند، می دانیم که اینها پارامترهای بسیار مناسبی برای درمان خواص آلیاژی هستند. علاوه بر این، تأکید می شود که این دو پارامتر نشان دهنده ویژگی جدول تناوبی دو بعدی است. استفاده از تنها یک پارامتر (به عنوان مثال، شماره مندلیف) هیچگاه تغییر خاصیت آلیاژ را در جدول تناوبی بیان نمی کند.
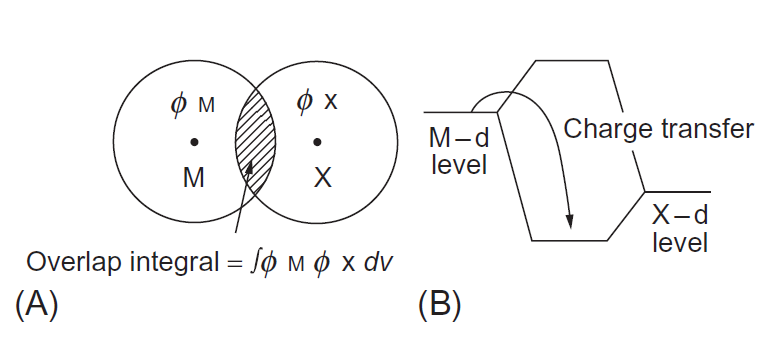
شکل 1-3-1 پارامترهای آلیاژی، (A) ترتیب پیوند، Bo و (B) سطح مدار d، Md. در (A)، ФM و ФX مدارهای اتمی M و X هستند و Bo متناسب با همپوشانی بین آنهاست.
محاسبه مدار مولکولی برای آلیاژهای نیکل
این روش برای اولین بار بر روی سوپرآلیاژهای پایه نیکل، که در آن فاز Ni3Al (γ΄) در ماتریس Ni (γ) رسوب میکند، اعمال شد. فاز Ni3Al (γ΄) یک فاز تقویت کننده است و کسر حجمی آن به میزان 60٪ در سوپرآلیاژهای پیشرفته بر پایهی نیکل است، که معمولاً برای موتورهای جت و توربین های گازی استفاده می شود.
همانطور که در شکل 1-3-2A نشان داده شده است، Ni3Al دارای یک ساختار از نوع L12 است [8]. با قرار دادن یک اتم Al در مرکز، مدل خوشهای همانطور که در شکل 1-3-2B نشان داده شده است ساخته می شود. این شامل یک اتم مرکزی Al، 12 اتم Ni در نزدیکترین همسایگی و 6 اتم Al در دومین همسایگی است. یک عنصر آلیاژی، M، برای یک اتم Al مرکزی جایگزین می شود. بنابراین، مدل خوشهای به صورت (MNi12Al6) بیان میشود، و فلزات انتقال d3، d4 و d5 برای M انتخاب می شوند. از آنجایی که M با 12 اتم Ni احاطه شده است، M در شرایطی مانند fcc Ni قرار میگیرد. فواصل بین اتمی از پارامتر شبکه اندازه گیری شده تنظیم می شود. چگالی حالت الکترونها برای Ni3Al خالص از محاسبه خوشه بدست میآید. ویژگی های اصلی نتایج حاصل از محاسبه باند است، به رغم این که یک مدل خوشهای نسبتاً کوچک در محاسبه حاضر بکار رفته است [8].
ساختار سطح محاسبه شده در شکل 1-3-3 نشان داده شده است، جایی که سطح انرژی فرمی، Ef، مربوط به Ni3Al صفر تنظیم شده و به عنوان مرجع مورد استفاده قرار میگیرد. برای یک خوشه خالص Ni3Al، سطح g1a13 تا eg15 عمدتا از مدار Ni 3d به دست میآید و باند Ni 3d در جایی که Ef قرار دارد تشکیل میشود، همانطور که توسط یک فلش نشان داده شده است. برای خوشه آلیاژ شده با فلزات انتقال d3 (M)، سطح انرژی جدید که عمدتا از مدارهای M-d سرچشمه میگیرد، در بالای Ef مشاهده می شود. به عنوان مثال، آنها سطح eg16 و g2t14 هستند که توسط خطوط نقطه دار در شکل ترسیم شده اند. ارتفاع آنها به طور یکنواخت با عدد اتمی M کاهش می یابد. میانگین مقدار این دو سطح به عنوان سطح Md برای M تعریف می شود. همچنین، ترتیب پیوند، Bo، بین الکترون های Ni-d و Md از M های مختلفی از آنالیز جمعیت Mulliken بدست می آید [11].
مقادیر محاسبه شده Md و Bo در جدول 1-3-1 آورده شدهاند. براساس ترتیب پیوند، درگروه A6، عناصر Cr، Mo و W بزرگترین انرژی پیوند را نشان میدهند. لازم به ذکر است که عناصر Cr، Mo، Ta، W و Re که دارای Bo بالایی هستند، جز عناصر آلیاژی در آلیاژهای پایه Ni میباشند]10[. بنابراین، Bo یکی از ملکهای تعیین عناصر آلیاژی است.
برای هر آلیاژ دلخواه، مقادیر متوسط Md و Bo با در نظر گرفتن میانگین ترکیب آلیاژ مورد نظر تعریف می شوند و به صورت ذیل بیان می شوند:

در اینجا، Xi کسر اتمی مؤلفه i در آلیاژ و (Md)i و (Bo)i مقادیر Md و Bo برای مؤلفه i هستند. بدیهی است که Md مرکز گرانش در پیوندهای تراز d آلیاژ میباشند، زیرا Md میانگین ترکیب تراز d هر عنصر آلیاژی در آلیاژ مورد نظر است. اگر ترکیب آلیاژ شناخته شده باشد، هر دو مقادیرMd و Bo بهراحتی با استفاده از معادلات (1-3-1) و (1-3-2) محاسبه میشوند.
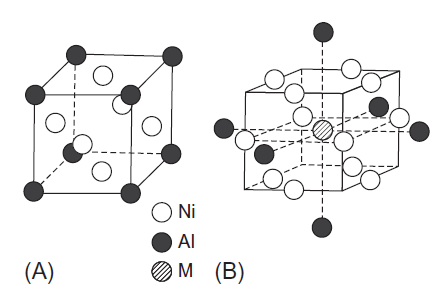
شکل 1-3-2 (A) ساختار کریستالی مدل خوشه Ni3Al و (B) مورد استفاده در محاسبه
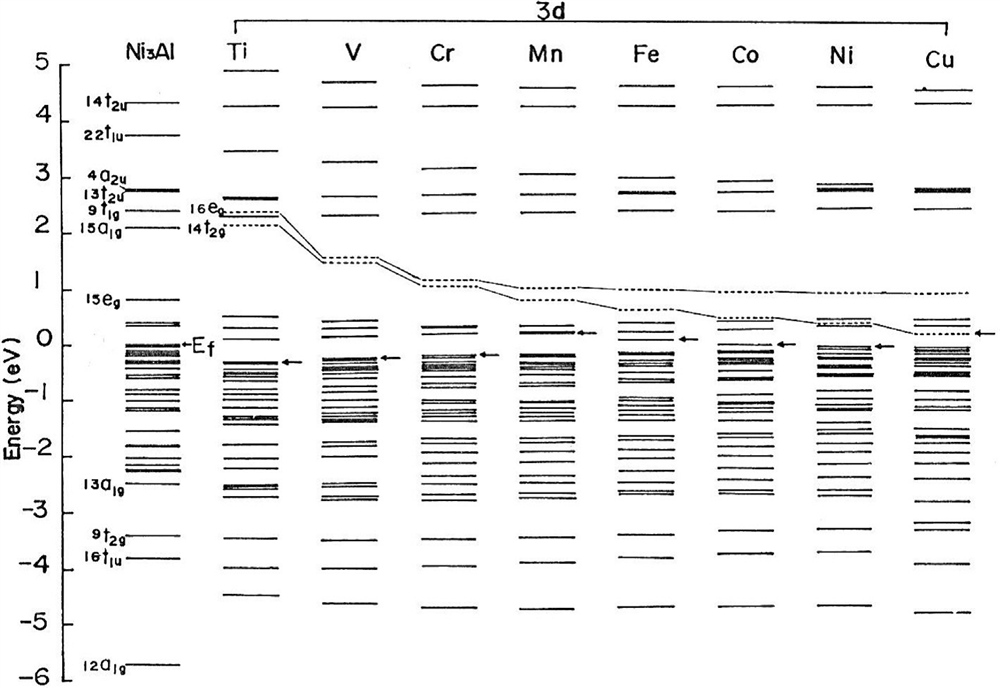
شکل 1-3-3 ساختارهای سطح انرژی Ni3Al خالص و آلیاژی با عناصر انتقال 3 بعدی. 16eg و 14t2g سطح d- مداری عنصر آلیاژی، M هستند.
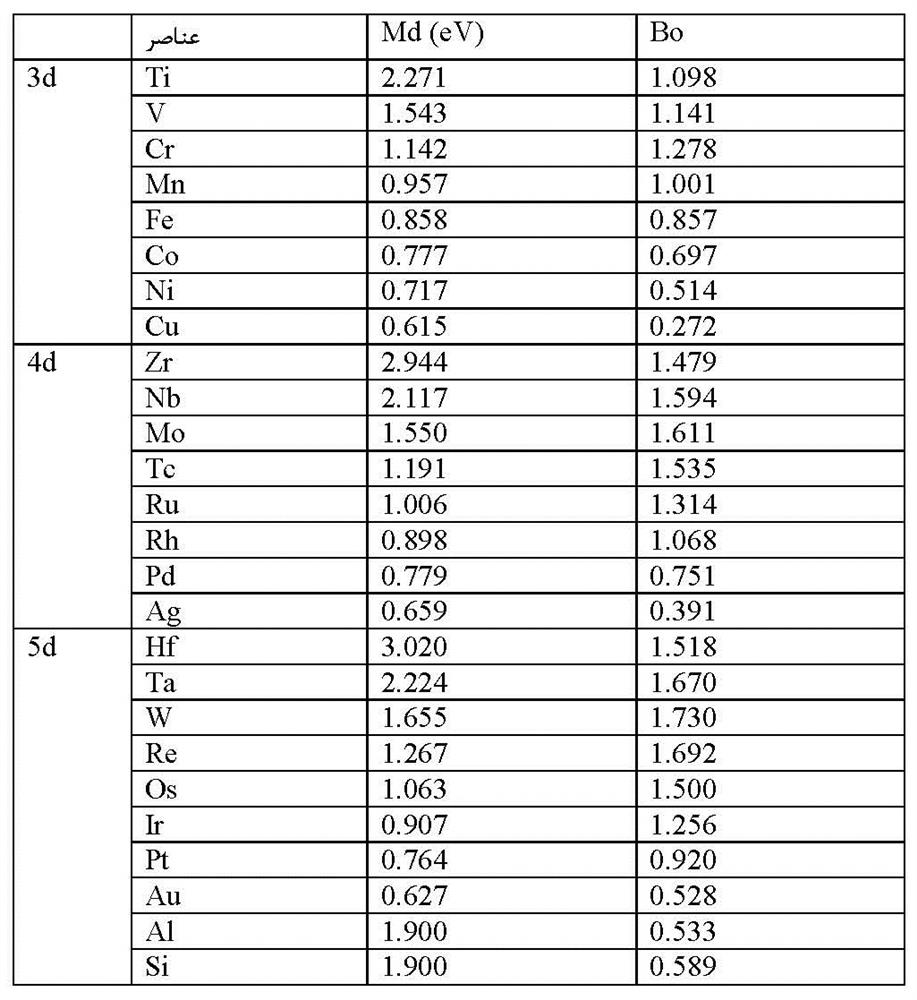
جدول 1-3-1 لیست مقادیر Md و Bo برای عناصر آلیاژی مختلف در نیکل
PHPCOMP جدید
در متالورژی فیزیکی، مشکل حلالیت بسیار مهم است. در رویکرد کلاسیک که توسط Hume-Rothy و Darken-Gurry ارائه شده است ]4،5[، این مشکل با استفاده از شعاع اتمی و الکترونگاتیوی حل شده است. ولی زمانی که اتمهای حلال و محلول هردو از جنس فلزات انتقالی باشند، این مشکل ناشناخته باقی میماند.
در سال 1964، روش PHACOMP (PHAse Computation) [1،2] در ایالات متحده گسترش یافت. این کار با هدف پیش بینی حدت حلالیت جامد عناصر در زمینه Ni (γ) و جلوگیری از تشکیل فازهای شکننده (به عنوان مثال، فاز σ) در ماتریس γ، مورد استفاده قرار گرفت. برای این روش، تعداد جاهایخالی الکترونیNv، استفاده می شود. در اینجا، Nv تعداد جاهایخالی الکترونی یا حفرههایی است که در بالای تراز انرژی فرمی در لایه پیوندی d قرار دارد، و بهطور تقریبی برابر با میباشد، که در این رابطه e/a نسبت الکترون به اتم میباشد. به عنوان مثال، برای عناصر گروه 4A از جمله Cr، Mo و W (که در آنها مقدار 4e/a= است)، مقدار Nv برابر با 66/6 میباشد.از آنجایی که Cr دارای شعاع اتمی کمتری نسبت به Mo یا W میباشد، بنابراین پارامتر Nv در اینجا مطابق با مفهوم اندازه اتمی قرار نگرفته است. بنابراین این روش مغایر با رویکرد کلاسیک توسط Hume-Rothy و Darken-Gurry [4،5] بوده و به ناچار پیش بینی Nv با این روش، ضعیف میباشد. با این وجود، PHACOMP در سراسر جهان برای طراحی و کنترل کیفیت سوپرآلیاژهای پایه Ni مورد استفاده قرار گرفته است.
نویسنده یک PHACOMP جدیدی را در سال 1984 پیشنهاد کرد [9]. در این روش، بهجای پارامتر Nv، از پارامتر Md برای پیش بینی حد حلالیت جامد استفاده میشود. همانطور که قبلاً ذکر شد، پارامتر Md برای اولینبار برای یک خوشهی آلیاژی که دارای موقعیت شیمیایی یکسانی در آلیاژ پایه Ni با ساختار FCC میباشد، محاسبه شده است. همچنین پارامتر Md دارای رابطه نزدیکی با الکترونگاتیوی و شعاع اتمی میباشد. به همین منظور، پارامتر Md پتانسیل برای پیش بینی حد حلالیت جامد آلیاژها از جمله آلیاژهایی که در آنها هردو اتم محلول و حلال از فلزات انتقالی بوده را دارا میباشد.
دو دیاگرام فازی معمولی در دمای K1477، در شکلهای A1-3-4 که مربوط به Ni-Co-Cr و شکل B1-3-4که مربوط به Ni-Cr-Mo بوده، نشان داده شده است، که در آن مرز فازی γ/γ + σ با استفاده از خط iso-Md و برابر با 925/0، نشان داده شده است. به منظور سادگی،مقدار واحد (ev) Md در شکل حذف شده است. به منظور مقایسه ، هر دو خط iso-Nv و خط iso-R در هر دو دیاگرام فازی ترسیم شدهاند. در اینجا، Nv و R به ترتیب میانگین ترکیبی NV و شعاع اتمیR، عناصر موجود در آلیاژ هستند. خط iso-NV از Nv=2.49، که اغلب برای پیش بینی مرز فازی γ / γ + σ استفاده می شود، بسیار دور از مرز است. از سوی دیگر، خط iso-R نزدیک به مرز نشان داده در شکل (A) است، اما از مرز نشان داده شده در شکل(B) بسیار فاصله دارد. در مقایسه با این موارد، خط Md iso- در هر دو شکلهای (A) و (B) به مرز نزدیک است. اعتبار این روش Md از طریق یکسری اندازهگیریها برای بیش از 30 دیاگرام فازی سهتایی، تأیید شده است [16-13]. ممکن است با کمک روش Md، برخی ابهامات مربوط به دیاگرامهای فازی آزمایشی را پیدا کنیم [16-13].
علاوهبر دیاگرامهای فازی سهتایی، آلیاژهای تجاری چند جزئی در دسترس، با این روش Md قابل حل هستند [9]. PHACOMP جدید با موفقیت در طراحی سوپرآلیاژهای پایه Ni، اعمال شده است [20-17].
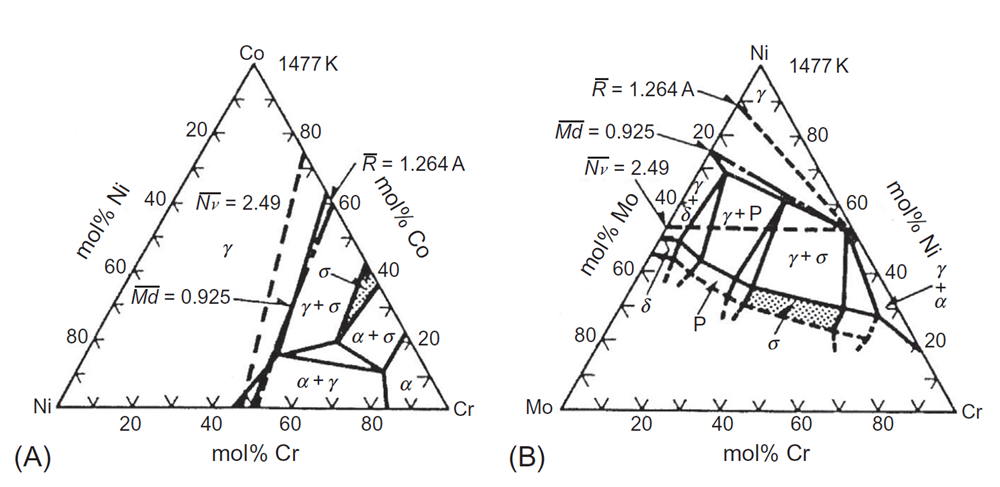
شکل 4-3-1 پیش بینی مرز فازی γ / γ + σ در دیاگرامهای فازی سهتایی(A) Ni-Co-Cr و(B) Ni-Cr-Mo.
محاسبه اوربیتال مولکولی و پارامترهای آلیاژ آلیاژهای تیتانیوم
محاسبه اوربیتال مولکولی
در محدوده دمایی K1155برای Ti خالص، تحول آلوتروپیک بین hcp Ti (α-Ti) و bcc Ti (β-Ti) اتفاق میافتد. هر دو مدل خوشه ای bcc و hcp برای محاسبه ساختار الکترونیکی استفاده شدهاند[21]. به عنوان مثال، مدل خوشهای bcc در شکل A1-3-5 نشان داده شده است. مطابق با شکل، این ماده از یک اتم Ti در مرکز، هشت اتم Ti در نزدیکترین همسایه و شش اتم Ti در دومین همسایه تشکیل شده است که یک عنصر آلیاژی M، در مکان اتم Ti مرکزی جایگزین شده است. بنابراین، مدل خوشهای bcc با عنوان MTi14 بیان شده است. پارامتر شبکه مورد استفاده 0.3320 نانومتر است.
از طرف دیگر، مدل خوشهای hcp در شکل B1-3-5 نشان داده شده است. مطابق با شکل، این مدل شامل یک اتم Ti در مرکز، 12 اتم Ti در نزدیکترین همسایه و 6 اتم Ti در دومین همسایه میباشد که یک عنصر آلیاژی M، در مکان اتم Ti مرکزی جایگزین شده است. بنابراین، مدل خوشهای hcp با عنوان MTi18 بیان شده است. پارامترهای شبکه مورد استفاده nm 295/0 a=و nm 4683/0c= میباشند.
با استفاده از این مدلهای خوشهای، یکسری محاسبات برای عناصر آلیاژی مختلف M، برای تعیین پارامترهای آلیاژی مرتبط با تیتانیوم انجام شده است.
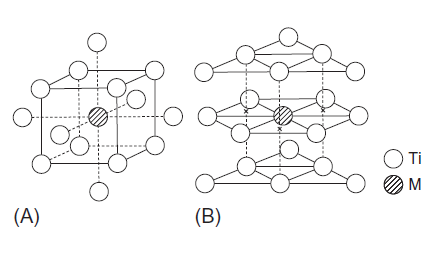
شکل 5-3-1 مدلهای خوشهای که برای محاسبات استفاده شدهاند، (A) خوشه bcc و (B) خوشه hcp.
پارامترهای آلیاژ
در شکل 6-3-1، ساختار تراز انرژی عناصر آلیاژی 3d در Ti با ساختار BCC نشان داده شده است ]21،22[. به عنوان مثال، نتیجهی محاسبات برای Ti خالص با استفاده از مدل خوشهای Ti15 (M=Ti) در سمت چپ تصویر نشان داده شده است که در آن تراز انرژی فرمی Ef با علامت ( ) مشخص شده است. ترازهای 18t1u تا 5eu بهطور عمده از اوربیتالهای 3d اتم Ti حاصل شدهاند، و پیوند 3d اتم Ti را در مکان Ef قرار میدهد. ترازهای انرژی پایینتر (از 17t1u تا 15a1g ) و ترازهای انرژی بالاتر (از 13eg تا 8t1g ) از پیوندهای 3d اتم Ti شامل 4s و 4p، تشکیل شده است. دو تراز انرژی، 16t2g و 13eg، که در حدود 2.5ev هستند، سطوح d اتم Ti مرکزی در مدل خوشهای نشان داده شده در شکل A1-3-5میباشند. کسر جزئی d در حدود 40٪ برای 16t2g و 59٪ برای 13eg میباشد.
در BCC Ti با عناصر آلیاژی 3d، تراز انرژی ناشی از اوربیتالهای M-d، در بالای EF ظاهر شدهاند. این سطوح اوربیتالی M-d همان ترازهای eg و t2g هستند که خطوط نقطهچین در شکل 6-3-1 نشان داده شدهاند، و شکل ظاهری آنها بسیار شبیه به نتایج مربوط به آلیاژ Ni3Al نشان داده در شکل 3-3-1 است. ارتفاع این سطوح با تعداد اتمی M، بهطور پیوسته کاهش یافته است. مقدار متوسط این دو سطح به عنوان Md برای آلیاژهای Ti تعریف شده است.
یکی دیگر از پارامترهای آلیاژی، ترتیب پیوند (Bo) است. ترتیب پیوند مرتبط با پیوند کووالانسی d-d، با استفاده از روش جمعیت Mulliken محاسبه شده است. نتایج در شکل A1-3-7 برای BCC Ti و برای Ti HCP در شکل A1-3-8 نشان داده شده است. در هر شکل، M-Ti و Ti-Ti ترتیب پیوند بین الکترونهای M-d و Ti-3d و بین الکترونهای Ti-3d و Ti-3d در یک خوشه است که مقدار کلی برابر با مجموع آنها است. ترتیب پیوند M-Ti بسته به میزان عنصر آلیاژی M، در هر دو نوع BCC و Ti HCP متغیر است. یک پیک کوچک در نزدیکی V وجود دارد و پس از آن با ترتیب عناصر کاهش مییابد، ترتیب پیوند کلی با رفتاری مشابه با ترتیب پیوند M-Ti، تغییر میکند. اندازه ترتیب پیوند کلی در عناصر 3d نسبت به عناصر 4d و 5d کمتر میباشد. روند تغییر ترتیب پیوند با M مشابه با رفتار مابین bcc Ti و hcp Ti است.
همانطور که در سوپرآلیاژهای پایه نیکل شرح داده شده است [10]، عناصر دارای ترتیب پیوند بالا، عناصر آلیاژی پایهای برای آلیاژهای با کاربردهای سازهای هستند. این مورد در آلیاژهای تیتانیوم نیز وجود دارد، زیرا عناصر آلیاژی اصلی مانند V، Cr، Zr، Nb و Mo از ترتیب پیوند بالایی برخوردار هستند. بهمنظور نشان دادن چگالی و استحکام خاص آلیاژها، ترتیب پیوند هر عنصر با وزن اتمی آن تقسیم میشود. نتایج در شکل B1-3-7 برای bcc Ti و در شکل B1-3-8 برای hcp Ti آورده شده است. مشخص است که شکل B1-3-7بسیار شبیه به شکل B1-3-8است. نسبت ترتیب پیوند به وزن اتمی برای Al، (Ti) و V بسیار زیاد است. از اینرو این نتیجه گرفته میشود که انتخاب عناصر آلیاژی در سیستم Ti-6Al-4V بسیار منطقی است.
در جدول 2-3-1، پارامترهای Bo و Md برای عناصر آلیاژی مختلفM ، در bcc Ti ذکر شده است. پس از آن، پارامترهای به دست آمده برای bcc Ti برای آنالیزهای پیشرو به عنوان خصوصیات آلیاژ استفاده شدهاند، زیرا به نظر میرسد که این پارامترها نسبت به ساختار کریستالی نسبتاً غیر حساس هستند [21،22].
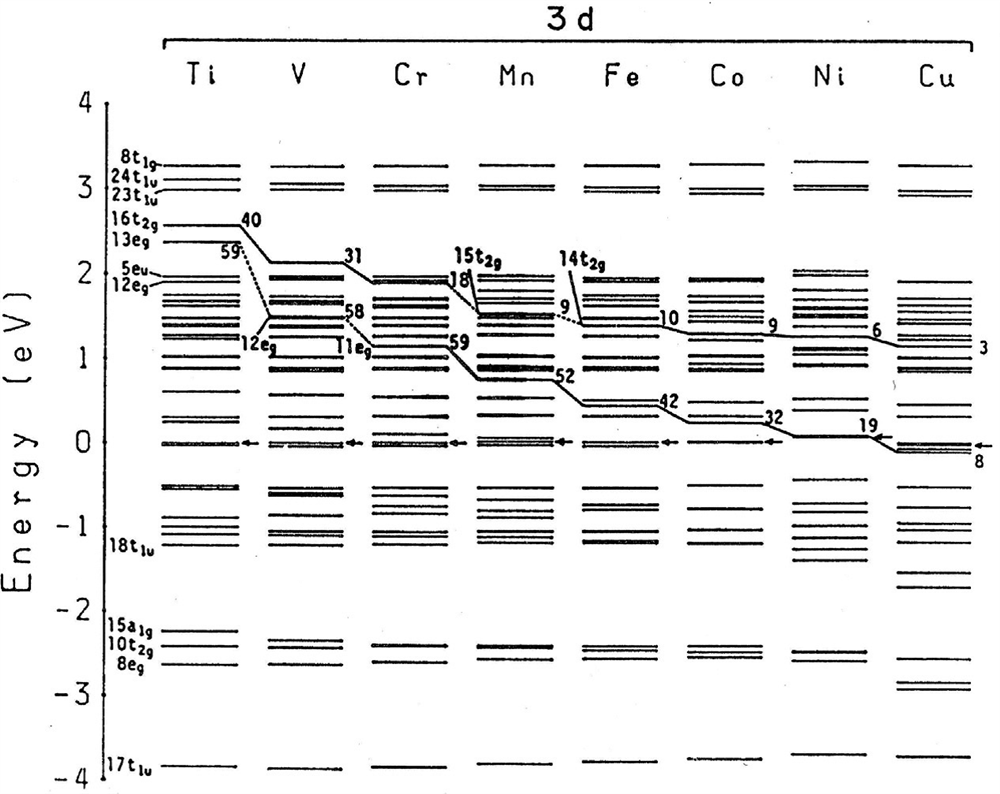
شکل 6-3-1 ساختار سطح انرژی bcc Ti حاوی عناصر انتقالی 3d.
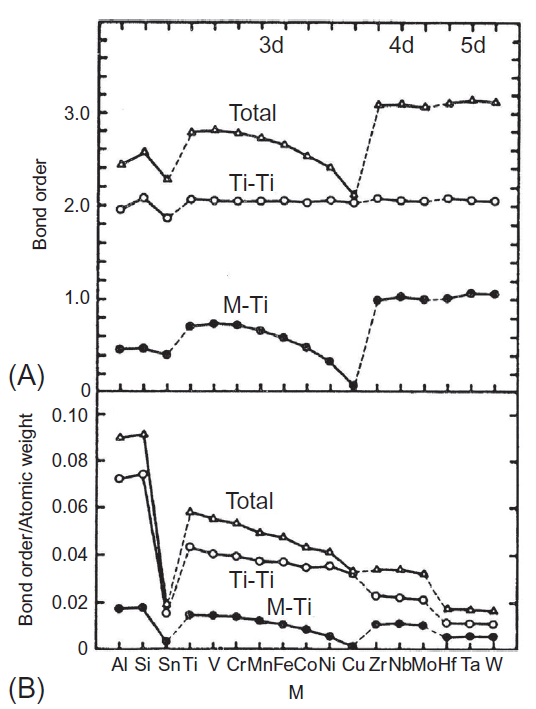
شکل 7-3-1 (A) ترتیب پیوند و (B) ترتیب پیوند به وزن اتمی برای M در bcc Ti.
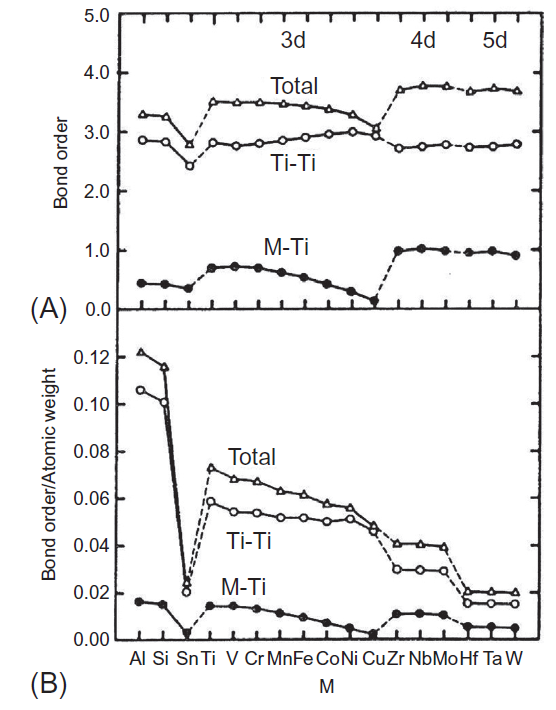
شکل 8-3-1 (A) ترتیب پیوند و (B) ترتیب پیوند به وزن اتمی برای M در hcp Ti.
ارتباط پارامترهای آلیاژی با خواص آلیاژ
دستهبندی دیاگرامهای فازی دوتایی
در شکل A-C1-3-9 سه دیاگرام فازی معمولی آلیاژهای دوتایی Ti-M نشان داده شده است، که M یک فلز انتقالی است. در اینجا، (A) نوع محلول جامد جامد کامل α و β، (Β) نوع β- ایزومورف و (C) نوع β-utectoid است. همانطور که در شکل 9-3-1 نشان داده شده است، این سه نوع دیاگرام فازی را میتوان به وضوح در نمودار Bo و Md تشخیص داد. دیاگرام فازی Ti-W قبلاً در (C) دستهبندی شده بود، اما طبق یک مطالعه اخیر، در گروه بندی آن تجدید نظر شده و در گروهبندی (B) قرار دادهاند ]23[.
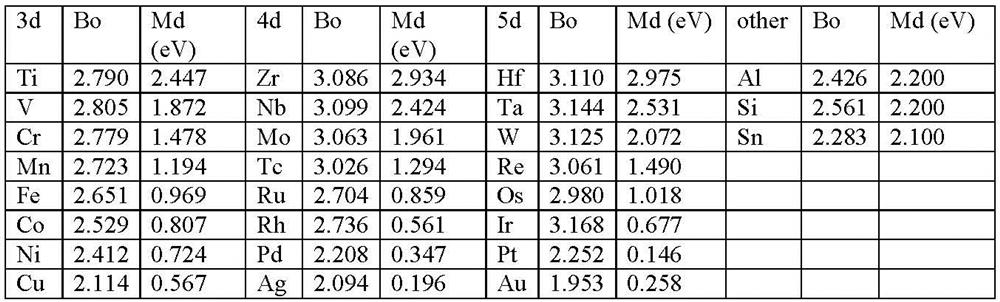
جدول 2-3-1 لیست مقادیر Md و Bo برای عناصر مختلف آلیاژی در bcc Ti
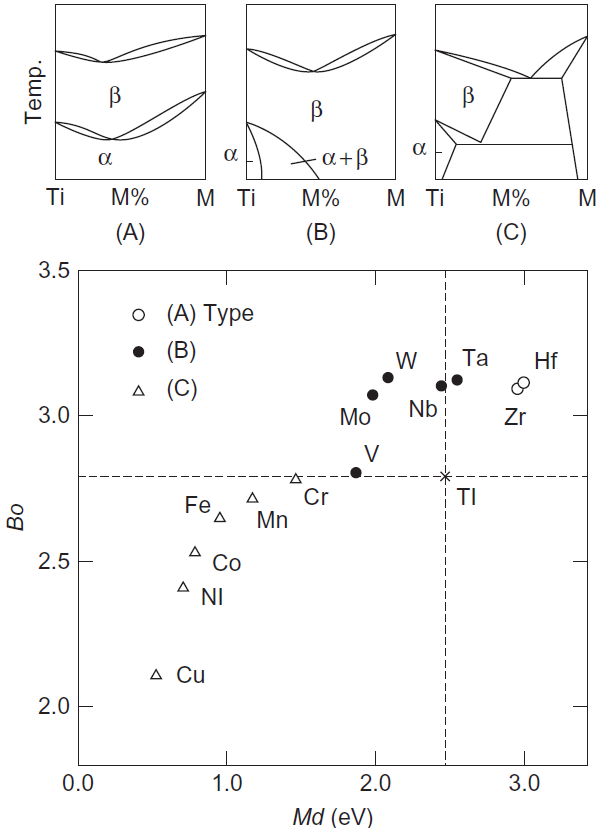
شکل 9-3-1 نمایش آلیاژهای دوتایی Ti-M در نمودار Bo در مقابل Md.
طبقه بندی آلیاژهای مورد استفاده در انواع α، α + β و β
ما معمولاً آلیاژهای تیتانیوم را از فازهای موجود در آلیاژ به انواع α، α + β و β دستهبندی میکنیم. حدود 40 آلیاژ بکار رفته در نمودارBo-Md در شکل 10-3-1 نشان داده شده است که مقادیر Bo و Md برای هر آلیاژ با استفاده از معادلات (1-3-1)، (2-3-1) محاسبه شدهاند [21،22]. در اینجا، ترکیبات آلیاژ با درصد جرمی مشخص شدهاند. در آلیاژهای دوتایی Ti-M، فاز β دما بالا، حتی در دمای اتاق وقتی که ترکیب آلیاژ از یک مقدار بحرانی خاص خارج شود، به طور قابل ملاحظهای بهصورت نیمه پایدار باقی میماند. این مقدار بحرانی با M تغییر میکند، و چنین دادههایی برای M های مختلف، برای M های مختلف در این دیاگرام (مانند (a) - (g)) ترسیم شدهاند.
از شکل 10-3-1 مشخص است که سه نوع آلیاژ به طور جداگانه در این دیاگرام قرار دارند. به عنوان مثال، آلیاژ Ti-6Al-4V (شماره 9) در قسمت مربوط به نوع α + β قرار دارد. همچنین، آلیاژ Ti-8Mn (شماره 6) با علیرغم این که آلیاژ نوع α + β است، در زمینه β قرار دارد. با این حال، این یک تناقض نیست زیرا همانطور که در شکل 10-3-1 نشان داده شده است، Ti-8Mn در واقع یک آلیاژ نوع β است، زیرا مقدار 8٪ منگنز در آلیاژ از مقدار بحرانی 6٪ منگنز بالاتر است. اما آلیاژ برای بهبود خاصیت مکانیکی، در محدوده دمای (α + β) تحت عملیات حرارتی قرار گرفته و اتفاقاً در آلیاژ نوع α + β دسته بندی میشود. حتی اگر نوع آلیاژ نامشخص باشد، با محاسبه مقادیر و از ترکیب و ترسیم محل آلیاژ روی دیاگرام Bo-Md که در شکل 10-3-1 نشان داده شده است، قابل پیش بینی میباشد و دیگر برای این پیشبینی نیازی به انجام آزمایش نمیباشد.
علاوهبر این، به منظور درک رفتار آلیاژی عناصر مربوطه در دیاگرام Bo-Md، بردار آلیاژی برای آلیاژهای دوتایی در شکل 11-3-1 ارائه شدهاند. بردار آلیاژی در موقعیت Ti خالص شروع شده و در موقعیت آلیاژ Ti-10mass%M خاتمه مییابد. با مقایسه شکلهای 10-3-1 و 11-3-1، به عنوان مثال ممکن است مشاهده کنیم که بردار آلیاژی برای آلیاژ Ti-Al با افزایش مقدار Al، وارد میدان فازی α شود، که این نشان دهنده آن است که Al یک عنصر پایدارکننده فاز α است. در مقابل آن، به عنوان مثال، عناصر مانند V، Nb و Ta تثبیت کننده β هستند، زیرا بردارهای آنها به سمت قسمت فاز β هدایت میشوند. این نتایج با رفتار آلیاژی شناخته شده عناصر موجود در تیتانیوم مطابقت دارد.
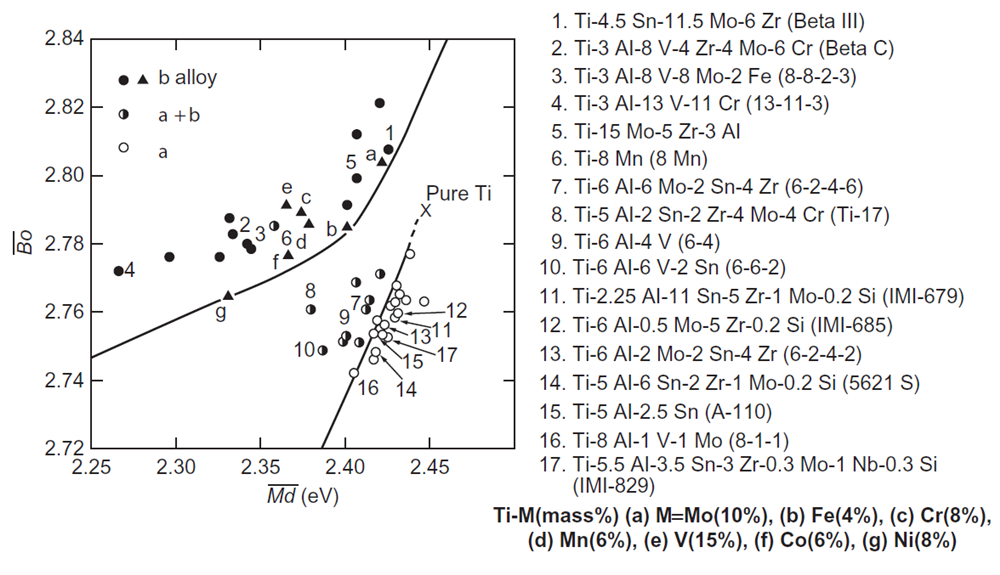
شکل 10-3-1 دستهبندی آلیاژهای تیتانیوم تجاری به سه نوع آلیاژهای α، α + β و β در نمودارBo-Md
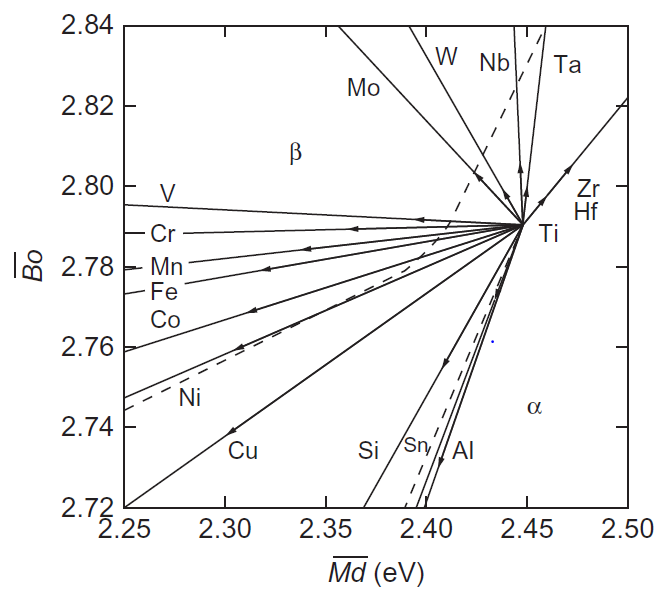
شکل 11-3-1 تغییر پایداری فاز با عناصر آلیاژی. بردار، محل آلیاژ Ti-10mass٪ M را نشان میدهد.
مرز بین لغزش و دوقلویی تغییرشکلی
در جریان تغییر شکل آلیاژهای نوع β، بسته به پایداری فاز β، هردو مکانیزم لغزش یا دوقلویی فعالیت دارند [24]. زمانی که پایداری فاز β بالا باشد، مکانیزم لغزش غالب بوده و زمانی که پایداری فاز β پایین باشد، مکانیزم دوقلویی مکانیزم غالب میباشد. ترکیب مرزی بین مکانیزم لغزش و دوقلویی با بررسی باندهای تغییر شکلی که در اطراف فرورفتگی ویکرز ظاهر شدهاند، تعیین میشود. همانطور که در شکل 12-3-1 نشان داده شده است، هنگامی که مکانیزم لغزش غالب است، باندهای لغزشی موجی ظاهر میشود، در حالی که باندهای دوقلویی مستقیم زمانی که مکانیزم دوقلویی غالب است، ظاهر میشوند. این مشاهدات برای انواع آلیاژهای دوتایی مانند Ti-V، Ti-Cr، Ti-Mn، Ti-Fe، Ti-Co، Ti-Nb، Ti-Mo، Ti-W و همچنین برای آلیاژهای چند جزئی Ti-Al-V-Cr-Mo-Zr با ترکیبات مختلف، انجام شده است. نتایج در دیاگرام Bo-Md نشان داده شده در شکل 12-3-1 خلاصه شده است. مشاهده شده است که بیشتر آلیاژهای تجاری موجود در امتداد ناحیهی مرزی لغزش/ دوقلویی قرار دارند. به عنوان مثال، آلیاژ Ti-10%V-2%Fe-3%Al (10-2-3) در منطقه مکانیزم دوقلویی بوده و آلیاژ Ti-15%V-3%Cr-3%Sn-3%Al (15-3-3-3) در منطقه مکانیزم لغزش در نزدیکی مرز لغزش/ دوقلویی قرار گرفته است.
علاوهبر ناحیهی تغییر شکلی لغزش یا دوقلویی، ناحیه ظاهر شده برای فاز مارتنزیت در این آلیاژ در دیاگرام Bo-Md شکل 12-3-1 مشخص شده است[22،24].
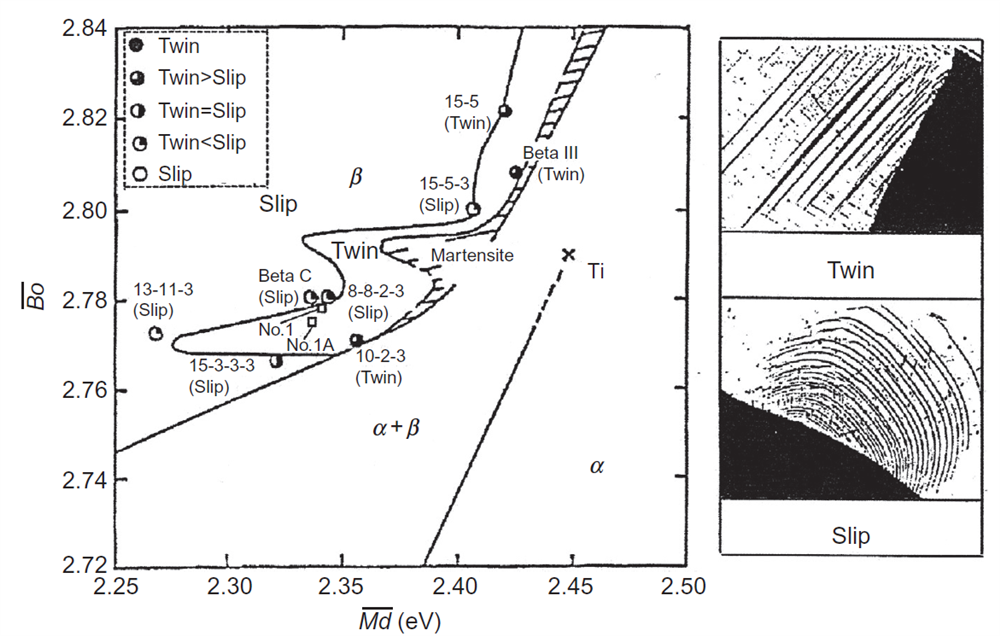
شکل 12-3-1 مرز لغزش/ دوقلویی و ناحیه مارتنزیت در دیاگرام Bo-Md
مقاومت در برابر خوردگی
مقاومت خوردگی بالا یکی از ویژگی های ویژهی تیتانیوم بوده، اما همچنان در اتمسفرهای احیایی همچن هیدروسورفوریک و هیدروکلریک اسید، خوردگی صورت میگیرد، اما مقاومت در برابر خوردگی تیتانیوم با آلیاژسازی بهبود مییابد. به منظور درک رفتار عناصر آلیاژی بر مقاومت خوردگی، یکسری از منحنیهای پلاریزاسیون در 343K در هر دو محلول 10mass% H2SO4 و 10mass%HCl، برای انواع آلیاژهای دوتایی Ti-M ( M=Al, V, Cr, Fe, Ni, Zr, Nb, Mo, Hf, Ta ) اندازهگیری شده است [25]. در شکلهای A و B 1-3-13 مشخص شده است که آلیاژهای تیتانیوم با مقادیر بالای Bo، چگالی جریان آندی بحرانی پایینی در منحنی پلاریزاسیون از خود نشان دادهاند، از اینرو حتی در محلول 10mass% H2SO4 در دمای 343K مقاومت خوردگی بالایی را نشان دادهاند. نتیجهی مشابهی نیز در محلول 10mass%HCl در دمای 343K بدست آمده است[25]. بنابراین، Bo پارامتر مناسب برای پیشبینی مقاومت خوردگی آلیاژهای Ti در محیطهای اسیدی مانند محلولهای H2SO4 و HCl است. از طرف دیگر، اگرچه اثر عناصر آلیاژی بر ایجاد لایه پسیو همچنان ناشناخته باقی مانده است، به خوبی شناخته مشاهده شده است که پسیو بودن باعث مقاومت خوردگی عالی آلیاژهای تیتانیوم می شود. موریشیتا و همکاران [26]، حالتهای الکترونیکی موضعی کاتد را با استفاده از روش خوشهای DV-Xα شبیه سازی کرده و مدل سلولی الکتروشیمیایی موضعی را ساختهاند که در شکل 14-3-1 نشان داده است. عناصر آلیاژی مورد بحث در اینجا، Re، Os، Ir، Pt، Au، Tc، Ru، Rh، Pd و M= Ag میباشند. همانطور که در شکل 2-3-1 نشان داده شده است، هر عنصر مقدار Md پایین تر از Ti را دارا میباشد، بنابراین انتقال بار در جهت Ti به M انجام میگیرد. بنابراین، در مدل سلولی نشان داده شده در شکل 14-3-1، ناحیهی زمینه تیتانیوم مانند آند و منطقه حاوی عنصر آلیاژی مانند کاتد رفتار میکند.
واکنش آندی در خوردگی محلولی به صورتTi2+ + 2e- Ti بیان شده و واکنش کاتدی به صورت 2H+ + 2e- H2 بیان می شود. زمانی که الکترونهای رسانش در ناحیهی کاتدی به صورت موضعی تجمع یابند، واکنش کاتدی رو به افزایش خواهد بود و بنابراین H+ به آرامی رو به تخلیه خواهد بود تا این که تغییرات هیدروژنی برروی سطح کاتدی فراهم شود. به دنبال این مدل پیشنهادی، تمایل به موضعی شدن الکترون از چگالی حالتهای نزدیک به تراز فرمی تخمین زده می شود، زیرا این یک اقدام برای نشان دادن ظرفیت الکترونهایی بوده که از منطقه کاتدی به منطقه آندی منتقل میشوند، همچنین تمایل به تخلیه الکترونها بهH+، به ارتفاع تراز فرمی وابسته میباشد. این امر به این دلیل است که تراز فرمی بالا، شرایطی را فراهم میکند که الکترونها به ناحیهی کاتدی پیوند ضعیفی داشته و بنابراین بهراحتی از سطح ناحیه کاتدی تخلیه میشوند.
این مدل پیشنهادی به صورت تجربی از اندازهگیری منحنیهای پلاریزاسیون انواع آلیاژهای Ti-0.1 mol%M تأیید شده است. به عنوان مثال، کاتدهای حاوی Ir، Pt، Ru، Rh و Pd دو شرط ذکرشده را برای فعال کردن واکنش کاتدی برآورده میکند. این بدان دلیل است که، همانطور که در شکل 15-3-1 نشان داده شده است، برای همهی این عناصر، چگالی حالتهای نزدیک به تراز فرمی به اندازه کافی بالا است که می تواند موضعی شدن الکترون را ترویج کند، اگرچه خود تراز فرمی هم به اندازه کافی بالا است تا تخلیه آرام الکترونها را فراهم کند. همچنین به صورت تجربی مشاهده شده است که هر آلیاژ حاوی این عناصر، پتانسیل اضافی اندازهگیری شده کوچک بوده و منحنی پلاریزاسیون کاتدی به سمت پتانسیل خنثی تغییر میکند. این نتایج تجربی به وضوح نشان میدهد که واکنش کاتدی افزایش یافته است، که مطابق با پیشبینیهای تئوری میباشد. بنابراین، مدل پیشنهادی بر پایهی محاسبه اوربیتال مولکولی به منظور نمایش حالتهای الکترونی موضعی کاتد در محیطهای خورنده آبی، مناسب است.
با استفاده از رویکرد حاضر، اوزاکی و همکاران ]27[، آلیاژهای جدید α + β که نسبت به Ti-6Al-4V دارای مقاومت به خوردگی بالاتری را در اتمسفری اسیدی هستند را گسترش دادهاند. ترکیبات آلیاژی برای ایمپلنتهای پزشکی شامل Ti-15%Zr-4%Nb-2%Ta-0.2%Pd و Ti-15%Sn—4%Nb-2%Ta-0.2%Pd بوده که عاری از عناصر سمی V و Al میباشند.
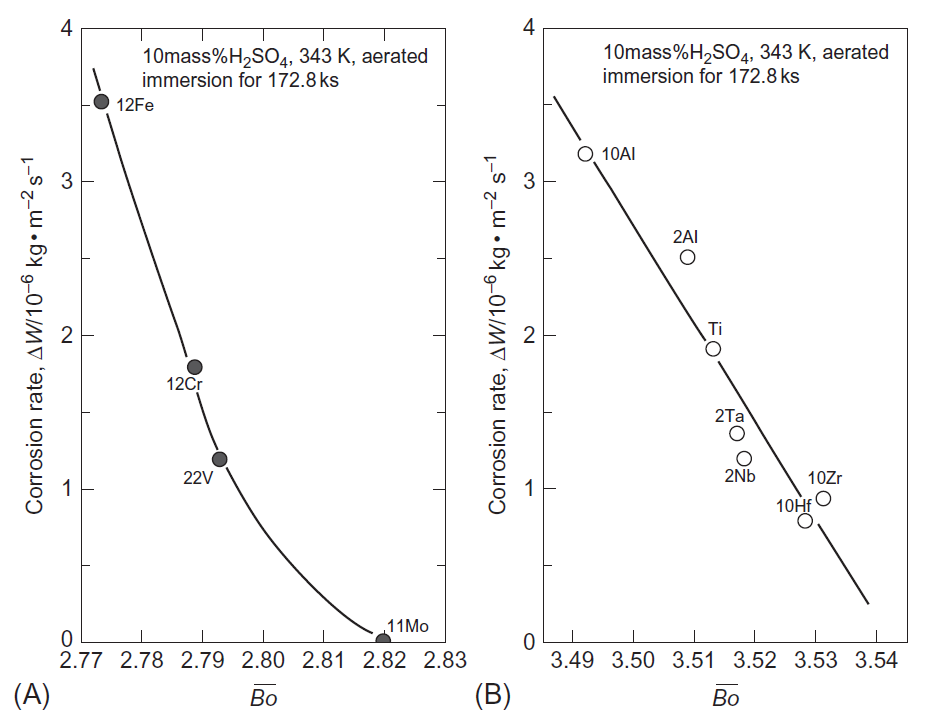
شکل 13-3-1 ارتباط نرخ خوردگی، ΔW، با Bo برای آلیاژهای دوتایی در 10%H2SO4 در دمای 343K برای (A) ساختار BCC و (B) ساختار HCP
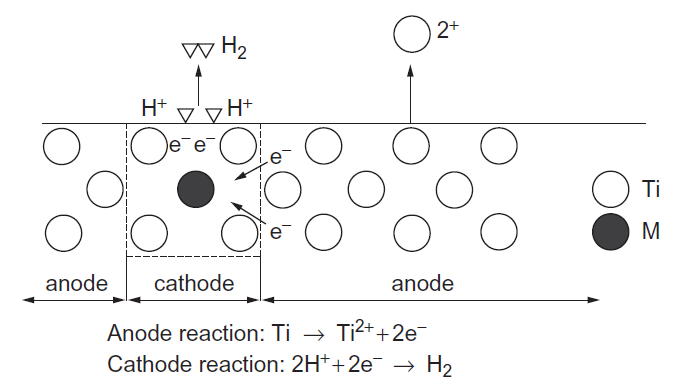
شکل 14-3-1 مدل شماتیک سلول الکتروشیمیایی.
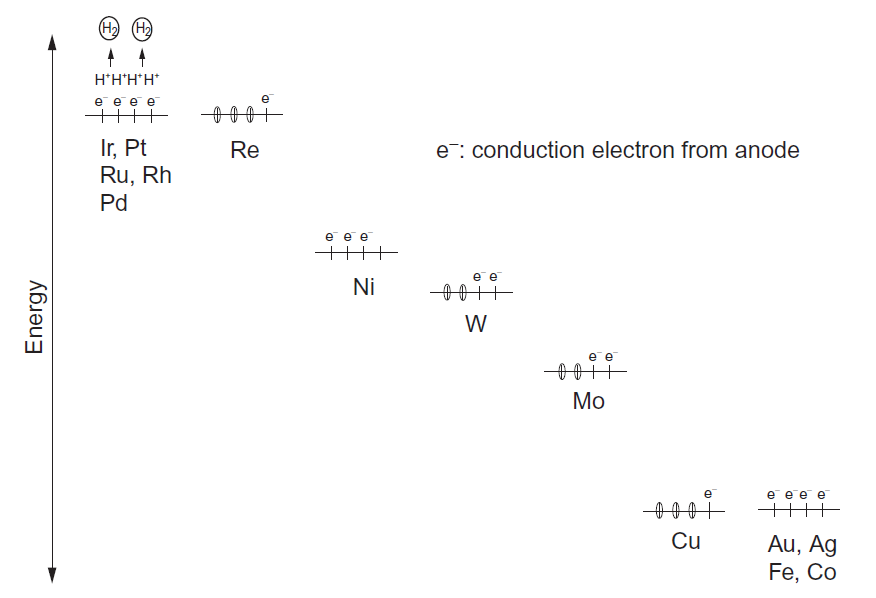
شکل 15-3-1 مدل الکترونیکی برای واکنش کاتدی.
طراحی آلیاژهای تیتانیوم
آلیاژهای استحکام بالای نوع β
ابتدا، یک مثال از طراحی آلیاژ بصورت بتن مشخص توضیح داده شده است. زمانی که یک سیستم آلیاژی مشخص با ناحیه Bo-Md خاص برروی نمودار نشان داده شده در شکل 10-3-1 تنظیم شود، ترکیب آلیاژ با اعمال جمع برداری بر بردارهای آلیاژی نشان داده شده در شکل 11-3-1، بدست میآید. در این حالت دیگر نیازی به بهینهسازی ترکیب آلیاژ از با استفاده از روش آزمایش و خطا نیست.
همانطور که در ابتدا شرح داده شد، Ti-15-3-3-3 با مکانیزم لغزش تغییر شکل مییابد اما Ti-10-2-3 با مکانیزم دوقلویی تغییر شکل مییابد. در حالت کلی، در آلیاژهای تغییر شکلی با دوقلویی نسبت به آلیاژهای تغییرشکل یافته با لغزش، تنش تسلیم کمتر و انعطافپذیری تا شکست طولانیتری انتظار میرود [24]. این بدان معنی است که دوقلوییهای تغییر شکلی، کارپذیری دما پایین (کارسرد) را افزایش داده در حالی که تغییر شکل انجام شده با لغزش، استحکام بالای آلیاژهای Ti را نتیجه میدهد. به منظور برطرف کردن این ماهیت متضاد بین کارپذیری دما پایین و استحکام بالا، ایدهی طراحی یک آلیاژ جدید که مکانیزم دوقلویی در حالت عملیات محلول سازی شده و مکانیزم لغزش در حالت پیر شده برقرار باشد، ارائه شده است [24]. به عنوان مثال، آلیاژ Ti محلول سازیشده که تحت نورد سرد قرار میگیرد، دارای کشیدگی شکست بالایی باشد ولی در حالت نهایی پیرسازی شده نیز دارای استحکام بالایی باشد تا شرایط سرویسدهی طولانی مدت این آلیاژ را تضمین کند.
مطابق دیاگرام Bo-Md، اگر آلیاژ در منطقه دوقلویی در شکل 12-3-1 قرار داشته باشد، تغییر شکل دوقلویی در حالت محلولسازیشده صورت میگیرد. با این حال، هنگامی که فاز α در آلیاژ نوع β با عملیات پیرسازی پیدیپی تجزیه می شود، ترکیب آلیاژ زمینه باقیمانده به ناحیهای از نمودار که β پایدارتر است، تغییر می یابد به طوری که به ناچار تغییر شکل لغزشی در حالت پیرسازی شده کار فعالیت میکند.
در این دیدگاه، آلیاژهای استحکام بالای از نوع β، Ti-13%V-3%Cr-2%Nb-92-30%Al (درصد جرمی) طراحی شده اند [24]. پس از این، آلیاژ حاوی 2٪ آلومینیوم، به عنوان آلیاژ شماره 1 و آلیاژ حاوی 3٪ آلومینیوم به عنوان آلیاژ شماره 1A نامگذاری شده است که موقعیت آنها در شکل 12-3-1 نشان داده شده است.
در طراحی این آلیاژها، V و Al به دلیل این که دارای نسبت ترتیب پیوند به درصد وزن اتمی بالایی هستند ( مطابق با شکلهای B1-3-7 و B1-3-8) انتخاب شدهاند و به همین دلیل استحکام ویژهی بالایی ممکن است. مشخص است که آلومینیوم یک عنصر استحکامی برای فاز α است β که با عملیات حرارتی پیرسازی مناسب درون زمینه β رسوب میکند. Cr به منظور بهبود انعطافپذیری آلیاژ انتخاب شده است. Nb نیز به منظور قراردادن موقعیت آلیاژ در ناحیهی بهتری از دیاگرام Bo-Md، انتخاب شده است. برای این سیستم آلیاژی Ti-V-Al-Cr-Nb، محل آلیاژ در داخل ناحیهی دوقلویی اما نزدیک به مرز لغزش/ دوقلویی انتخاب شده است که در شکل 12-3-1 نشان داده شده است. آلیاژهای شماره 1 و 1A در دیاگرام Bo-Md، نزدیک به ناحیهای مابین دو سیستم Ti-10-2-3 و Ti-15-3-3-3 قرار گرفتهاند.
نتایج آزمون کشش برای دو حالت محلول سازی شده و پیرسازی شده در جدول 3-3-1 آورده شده است. مشاهده میشود که در حالت محلول سازی شده و در دمای 1093K برای هر دو سیستم آلیاژی، مکانیزم دوقلویی فعالیت دارد.استحکام تسلیم (YS) و استحکام نهایی (UTS) به ترتیب حدود 700MPa و 800MPa با انعطافپذیری حدود 30% گزارش شده است. با این حال، مکانیزم لغزش هنگامی که آلیاژ شماره 1 به مدت 18ks و آلیاژ شماره 1A به مدت 28.8ks در دمای 733K پیرسازی شده باشند، فعالیت مکانیزم لغزش گزارش شده است. اندازه فاز α پراکنده شده در آلیاژهای شماره 1 و شماره1A در حدود 60 نانومتر بوده که بسیار کوچکتر از اندازه فاز α سوزنی مانند در Ti-15-3-3-3 با طول تقریباً 35/0 میکرومتر و عرض 05/0 میکرون، میباشد. همانطور که در جدول 3-3-1 گزارش شده است، UTS برای آلیاژهای شماره 1 و1A برابر با 1400-1500MPa و بسیار بالاتر از آلیاژ Ti-15-3-3-3 که حدود 1200MPa گزارش شده است، میباشد. دانسیته آلیاژی اندازهگیری شده برای آلیاژ شماره 1 و 1A به ترتیب برابر با 4.684 و 4.660 (g/cm3) بوده که در مقایسه با آلیاژ Ti-15-3-3-3 با دانسیته 4.718 (g/cm3) کمتر میباشند. (g / cm3) است. همچنین این دو آلیاژ نسبت به آلیاژ Ti-15-3-3-3 دارای چقرمگی شکست بالاتری میباشند ]24[. بنابراین، با این روش تئوری طراحی آلیاژ، آلیاژ جدید نوع β، با ترکیب Ti-13%V-3%Cr-2%Nb-(2-3)%Al ، بدون هیچگونه آزمون و خطایی توسعه یافتهاند.
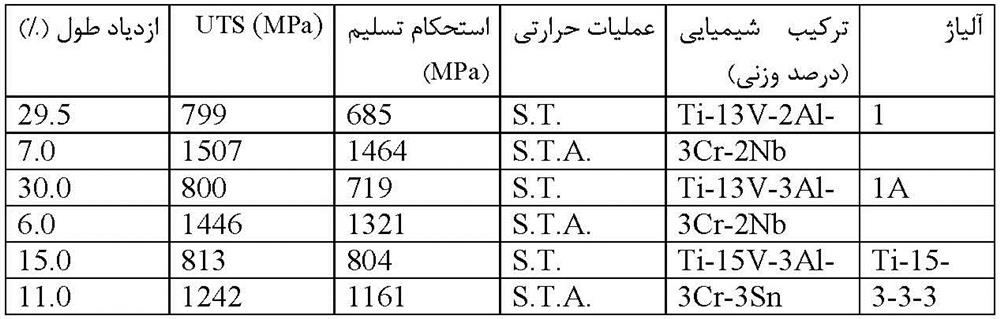
جدول 3-3-1 نتایج تجربی آزمون کششی برای آلیاژهای طراحی شده (شماره 1 و شماره 1A) و Ti-15-3-3-3
کاربردهای پزشکی آلیاژ نوع β
اخیراً، آلیاژهای نوع β به دلیل داشتن خواص منحصر بهفردی از جمله خاصیت حافظهداری، سوپرالاستیسیته، و مدول یانگ پایین توجه ویژهای در حوزهی پزشکی یافتهاند [28]. به عنوان مثال، برای جایگزین کردن استخوان انسان با یک آلیاژ، زمان و تلاش زیادی برای ساخت آن دسته از آلیاژهای نوع β که دارای مدول یانگ پایین و استحکام بالا هستند، صرف شده است. برای طراحی این دسته از آلیاژهای Ti با کاربرد پزشکی، از رویکرد اوربیتالی مولکولی استفاده کردهاند.
به عنوان مثال، نینامی و همکارانش [29] آلیاژ جدیدی از نوع β با ترکیب Ti-29%Nb-13%Ta-4.6%Zr (TNTZ) طراحی کردهاند که از عناصر غیرسمی مانند Nb، Ta و Zr استفاده شده است و دارای مدول یانگ حدود 50GPa بوده که نسبت به آلیاژ Ti-6Al-4V کمتر میباشد.
همچنین سایتو و همکارانش [30] گزارش کردهاند که فلزات لثهای (gum) در همسایگی آلیاژ TNTZ قرار دارند. یعنی، موقعیتهای آنها در دیاگرام Bo-Md، نزدیک به Bo=2.87 و Md=2.45 ، میباشد. معیار دیگر برای ظهور فلزات لثهای، مقدار e/a بوده، که برابر با 4.24 میباشد و در این حالت ترکیبات آلیاژی برابر با Ti-12%Ta-9%Nb-3%V-1.5%O و Ti-23%Nb-0.7%Ta-2%Zr-1.2%O میباشد. زمانی که این فلزات لثهای تحت 90% کارسرد قرار میگیرند، علاوهبر خصوصیات منحصر بهفردشان، خاصیت سوپرالاستیسیته و مدول یانگ پایین نیز مورد توجه قرار میگیرد به همین منظور از فلزات لثهای برای استحکام سیمهای دندانی استفاده میشود.
گسترش نمودار Bo - Md در منطقه بالاتر Bo
به تازگی، دیاگرام Bo-Md نشان داده شده در شکل 10-3-1 به مناطقی که مقدارBo بیتشر از 2.84 میباشد، گسترش یافته است، زیرا آلیاژهای ویژهی نوع β مانند TNTZ فلزات لثهای در این ناحیه نمایان شدهاند که این دیاگرام گسترش یافته در شکل 16-3-1 نشان داده شده است [31].
پایداری فاز β به ترکیب آلیاژ یا دما بستگی دارد. فاز مارتنزیت و فاز ω (امگا) در برخی از محدودههای ترکیبی آلیاژهای نوع β ظاهر میشوند. یک رابطه فازی اساسی در دیاگرام گسترش یافتهی Bo-Md در شکل 16-3-1 نشان داده شده است. در مورد استحاله مارتنزیتی، این تحول از دمای Ms شروع شده و در دمای Mf خاتمه مییابد، که هردوی این دماها تا حد زیادی بسته به ترکیب آلیاژ تغییر میکنند. بههمین منظور، منحنیهای همدمای iso-Ms بهصورت Ms=RT و Mf=RT در دیاگرام Bo-Md ترسیم شدهاند.
در حالت خاص، به منظور دستیابی به ساختار تک فاز β، آلیاژ با حداقل پایداری فاز β که دارای حداقل عناصر پایدارکننده فاز β میباشد، توسعه یافته است. این آلیاژ با حداقل پایداری فاز β در امتداد ناحیه باریک سمت چپ یا بالای مرز فاز β / β + ω قرار دارد، که در شکل 16-3-1 نشان داده شده است. همچنین مشاهد شده است که این آلیاژ دارای مدول یانگ پایینی میباشد.
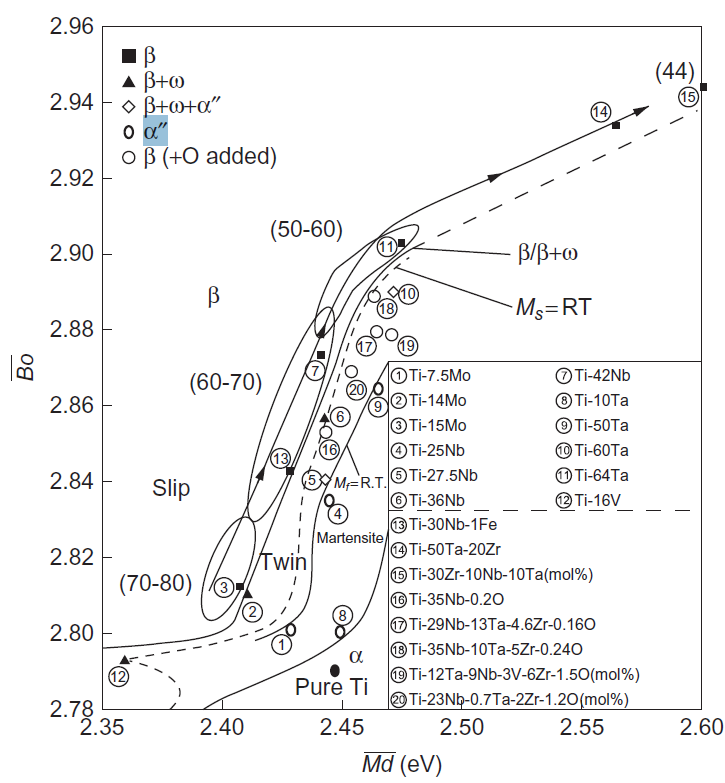
شکل 16-3-1 دیاگرام گسترش یافتهی Bo-Md به ناحیهای که Bo آن بالاتر از مقدار 2.84 است. مرز فازی β / β + ω همراه با مرز مارتنزیت برای خطوط همدمای Ms=RT و Mf=RT.
مدول یانگ بر حسب MPa برای برخی از آلیاژها مشخص شده است.
ارتباط پایداری فاز با خواص آلیاژ
پایداری فازی ارتباط نزدیکی به خواص آلیاژ از جمله مدول یانگ، سوپرالاستیسیته و خاصیت حافظهداری آلیاژ دارد. بنابراین، در اینجا از دیدگاه طراحی آلیاژ با استفاده از دیاگرام گسترش یافتهی Bo-Md به بررسی این موضوع پرداخته شده است [31].
مدول یانگ
مشخص شده است که فاز مارتنزیت α״ دارای مدول یانگ کمتری نسبت به فاز مارتنزیت α׳ است. فاز ω دارای بالاترین مدول یانگ است، بنابراین به منظور پایین نگهداشتن مدول یانگ، بایستی از رسوبگذاری آن جلوگیری شود [32]. در مقایسه با این فازها، فاز β کمترین مدول یانگ را در بیشتر آلیاژهای تیتانیوم دارا است [33،34]. همچنین، در آلیاژهای دوتایی Ti-M، پایداری فاز β با افزایش عناصر پایدارکننده β، افزایش مییابد. به عنوان مثال، هنگامی که عناصر آلیاژی V، Nb و W باشند، مدول یانگ اندازهگیری شده با افزایش درصد این عناصر بهطور یکنواخت، افزایش مییابد. به عبارت دیگر، همانطور که در بالا بیان شد، آلیاژ با حداقل پایداری فاز β، داری کمترین مدول یانگ در میان آلیاژها فاز β میباشد. به عنوان مثال، کمترین مدول یانگ در حدود 50MPa برای Ti-64Ta، 60 GPa برای Ti-40Nb و 72MPa برای Ti-26 V است، بنابراین تغییرات مدول یانگ به ترتیب آلیاژ برابر با Ti-Ta <Ti -Nb <Ti-V میباشد. همچنین یک نتیجهای برای کاهش مدول یانگ با افزایش مقدار Bo وجود دارد، زیرا برای Ti-26 V 2، Bo=2.794، برای Ti-26Nb، Bo=2.869 و برای Ti-64Ta ، برابر با Bo=2.903 میباشد.
این نتیجه از شکل 16-3-1 نیز نمایان است که در امتداد مرز فازی β / β + ω ، ناحیهای برای ساختارهای حداقل پایدار تک فاز β وجود دارد. در میان آلیاژهایی که با حداقل پایداری فاز β در امتداد این مرز فازی قرار گرفتهاند، همانطور که در شکل 16-3-1 نشان داده شده است، مدول یانگ با افزایش مقدار Bo ، کاهش مییابد. به عنوان مثال، با افزایش مقدار Bo، مدول یانگ از 70MPa در آلیاژ Ti-15Mo به 80MPa در آلیاژ Ti-30Zr-10Nb-10Ta تغییر مییابد. بنابراین، آلیاژکردن عناصر با مقدار Bo بالا مانند Zr، Nb، Mo، Hf، Ta و W برای طراحی آلیاژهای فاز β با کمترین مدول یانگ، مناسب میباشد.
تغییر در پایداری فاز β با افزودن عناصری مانند O، Al، Sn و Zr
افزودن مقدار کمی از عناصر پایدارکننده فاز α مانند O، Sn و Al به آلیاژهای فاز β باعث تغییر مرز فازی شده و خواص مکانیکی را تحت تأثیر قرار میدهند [30،32،35-38]. به عنوان مثال، گزارش شده است که افزودن O مدول یانگ را کاهش میدهد [35]. در آلیاژهای نوع β با مقادیر زیاد Bo، عناصر O، Sn و Al ، دیگر پایدارکننده فاز α نبوده ولی همانند عناصر پایدارکننده فاز β رفتار میکنند. به عنوان مثال، در آلیاژهای Ti-35Nb-7Zr-5Ta-(0.06-0.6)O [37]، شکل گیری فاز ω با افزودن O به تعویق میافتد، بنابراین O همانند عنصر پایدارکننده فاز β در آلیاژهای تکفاز β، عمل میکند. احتمالاً بهدلیل اثر قفلکنندگی O برروی نواقص خطی، مانع از فروپاشی صفحات اتمی {111} β در فاز β شده و بنابراین تشکیل فاز ω در این آلیاژها به تعویق میافتد. این ممکن است یکی از دلایلی باشد که چرا مقدار کمی O به آلیاژهای Ti که اخیراً توسعه یافتهاند، اضافه می شود [30،36،37،39،40]. همچنین اضافهکردن اکسیژن، مانع از تشکیل مارتنزیت میشود [41]. با این حال، اکسیژن هنوز هم مانند یک عنصر پایدارکننده فاز α در فرآیندهای پیرسازی عمل میکند، زیرا با افزایش مقدار O، مقدار فاز α رسوب یافته تحت فرآیند پیرسازی، افزایش مییابد [37].
در اینجا، ذکر شده است که هر نوع آلیاژ β از ⑯ تا ⑳ نشان داده شده در شکل 16-3-1 حاوی مقدار کمی اکسیژن است. با این حال، مقدار اکسیژن موجود در این آلیاژها در محاسبات لحاظ نمی شود، زیرا پارامترهای Bo و Md برای اکسیژن ناشناخته هستند. همانطور که در بالا بیان شد، اکسیژن از ویژگیهای عمومی عناصر پایدارکننده فاز α، پیروی نمیکند و بهجای آن همانند عناصر پایدارکننده فاز β رفتار میکند. بنابراین، با افزودن اکسیژن، همانطور که با نماد ● در شکل 16-3-1 نشان داده شده است، مرز تک فاز β به ناحیهای با مقدار عناصر کمتر پایدارکنند فاز β، تغییر مییابد. به عنوان مثال، برای آلیاژهای Ti-Nb-Ta-Zr-(V) و Ti-Nb، به نظر می رسد که افزودن O باعث گستردهتر شدن ناحیه تک فاز β شده که در شکل 16-3-1 با اعداد ⑯ تا ⑳ نشان داده شده است. به عبارت دیگر، با وجود این که این آلیاژها در واقع تک فاز β هستند، ولی اینها در منطقه β + ω (یا مارتنزیت) قرار داشته که در زیر مرز فاز β / β + ω میباشند. این در واقع محدودیتی برای این رویکرد است.
همچنین، زیرکونیوم (Zr) با توجه به پایداری فازی آن، به عنوان یک عنصر خنثی در نظر گرفته شده است. با این حال، تقریباً مانند اکسیژن موجود در آلیاژهای β رفتار میکند. یعنی، Zr مانند یک عنصر ضعیف پایدارکننده β در آلیاژ Ti-Nb-Zr [42] و Ti-Cr-Zr [38] رفتار میکند. با این حال اثر پایدارکنندگی فاز β آن در مقایسه با Al و Sn ضعیفتر است.
یک نمونه در شکل 17-3-1 برای آلیاژهای Ti-5Zr-pNb-10Ta-0.23O (p=20,25,30,35,40) و Ti-5Zr-30Nb-rTa-0.23O (r=0,5,10,15,20) نشان داده شده است [31،39]. از شکل 17-3-1 مشخص است که با افزایش مقدار Nb یا Ta، فاز β پایدارتر شده، بهطوری که تشکیل فاز ω در آنها به تعویق میافتد. همچنین، اضافه کردن همزمان عناصر O و Zr باعث انتقال مرزفازی β / β + ω به سمت پایینترین ناحیهی ترکیبی عناصر پایدارکننده β منتقل میکند که این ناحیه در شکل 17-3-1 با خطچین مشخص شده است. مشخصه مرزی آلیاژهای عاری از O و Zr با یک خاط پررنگ در شکل 17-3-1 نشان داده شده است. زمانی که این دو منحنی با یکدیگر مقایسه میشوند، بدیهی است که با آلیاژ کردن همزمان عناصر O و Zr،در دیاگرامBo-Md، مرز فازی تا حد زیادی به سمت آلیاژهای فاز β تغییر مییابد.اثر آلیاژی این عناصر جدید بر پایداری فاز β، به ترتیب برابر با O>Al>Sn>Zr، میباشد. همچنین با افزودن این عناصر به آلیاژهای فاز β، رفتار آلیاژی ویژهای پدید میآید [31].
اثر سوپرالاستیسیته و حافظهداری
برای آلیاژهای دوتایی Ti-Nb، سوپرالاستیسیته در آلیاژ با حداقل پایداری فاز β، مشخص شده است (به عنوان مثال، Ti-(40-20)Nb) [43]، که در آن تحول برگشتپذیر بین فاز β و مارتنزیت در دمای اتاق اتفاق میافتد، زیرا در این آلیاژهای با حداقل پایداری فاز β ،اختلاف انرژی بین فاز β و فاز α״ مارتنزیت بسیار ناچیز است. اثر حافظهداری در آلیاژهای با مقادیر کم Nb (به عنوان مثال، Ti-(35-40)Nb)، نمایان میشود [43]، که دمای Ms آنها نزدیک به دمای اتاق میباشد. با این حال، نه اثر سوپرالاستیسیته و نه اثر حافظهداری در آلیاژهای دارای فاز پایدار β (>45Nb)، که دمای Ms آنها کمتر از دمای اتاق میباشد، نمایان نمیشود. برای آلیاژهایی که مقادیر کمتری از Nb را دارا بوده و در ناحیهی مارتنزیتی قرار میگیرند (به عنوان مثال، Ti-30Nb)، هیچ یک از دو خاصیت سوپرالاستیسیته و حافظهداری نمایان نمیشود. با این حال، همانطور که در آلیاژ شماره ⑬ در شکل 16-3-1 نشان داده شده است، افزودن 1%Fe به آلیاژ Ti-30Nb، موقعیت این آلیاژ را به سمت آلیاژهای با حداقل فاز پایدار β منتقل میکند، بنابراین باعث نمایان شدن خاصیت سوپرالاستیسیته در آلیاژ میشود.
گسترهی ترکیبی از آلیاژهایی که هردو رفتار سوپرالاستیسته و حافظهداری را از خود نشان میدهند، با افزودن عناصر O، Al، Sn و Zr، بهبود مییابند. به عنوان مثال، همانطور که در بالا بیان شد، اثر حافظهداری در آلیاژ Ti-38.5Nb نمایان میشود، اما افزودن 1.5% Al به این آلیاژ، کرنش سوپرالاستیک 4.3% نتیجه میشود [44]. چنین تغییراتی در خواص آلیاژی، در آلیاژ Ti-36Nb با تغییر مقدار O مشاهده شده است. یعنی، اثر حافظهداری همچنان در آلیاژهای Ti-36Nb- (0-0.12) O نمایان میشود اما اضافهکردن بیشتر O باعث ایجاد خاصیت سوپرالاستیسیتی در آلیاژهای Ti-36Nb- (0.22-0.43) O میشود، که دارای فاز β با حداقل پایداری در دمای اتاق میباشند [36]. بنابراین، نمایان شدن اثر سوپرالاستیسیتی در آلیاژهای Ti-Nb حاوی O نسبت به آلیاژهای Ti-Nb فاقد O، بیشتر بهبود مییابد[36].
این امر در آلیاژهای چند جزئی Ti-5Zr-pNb-30Ta-0.23O (p=20,25,30,35,40) نیز صادق است [39]. هیچ کدام از دو پدیده در آلیاژهای فاز پایدار β (یعنی 35Nb)، مشاهده نشده است. با این حال، همانطور که در شکل 17-3-1 نشان داده شده است، خاصیت سوپرالاستیسیتی در آلیاژهای دارای حداقل پایداری فاز β (یعنی 30Nb) نمایان میشود. استحاله مارتنزیتی القاء شده توسط تنش، که مرتبط با اثر حافظهداری میباشد، در آلیاژهای حاوی مقادیر خیلی کم از Nb (یعنی 20Nb و 25Nb) مشاهده شده است [40]. در اینجا همانطور که قبلاً بیان شد، آلیاژ با حداقل پایداری فاز β (به عنوان مثال، 30Nb) پایینترین مدول یانگ را دارا میباشند.
بنابراین، پایداری فاز β آلیاژها با خواصی همچون مدول یانگ پایین، سوپرالاستیسیتی و اثر حافظهداری، ارتباط شدیدی داشته و چنین ارتباطی در دیاگرام Bo-Md، قابل مشاهده است. آلیاژسازی با عناصر ویژهای مانند O، Al، Sn و Zr، باعث تغییر مرز فازی بهطور قابل ملاحظهای میشود که زمینه اصلاحات فراوانی در پایداری فازی و از این رو در خواص آلیاژی را به روشی معقول فراهم میسازد.
نتیجه گیری
تئوری طراحی آلیاژ نه تنها برای درک اساسی آلیاژها، بلکه برای طراحی آلیاژ کاربردی بدون انجام آزمایشهای آزمون و خطا طراحی شده است [45]. بهویژه، این تئوری برای طراحی آلیاژهای Ti مناسب است، زیرا دیاگرام Bo-Md، نمایانگر ترکیبات فازی در آلیاژها است. چنین دانشی در مورد پایداری فازی در دیاگرام Bo-Md ارائه شده است، در پیشبینی خواص مختلف آلیاژ از جمله استحکام مکانیکی، خواص خوردگی، مدول یانگ، سوپرالاستیسیتی و اثر حافظهداری، مهم میباشد. با یادآوری اینکه همه این خصوصیات برای آلیاژهای Ti با کاربرد پزشکی ضروری است، بسیار انتظار می رود که این تئوری در عمل در زمینهی طراحی آلیاژ برای کاربردهای بیوتکنولوژی به خوبی عمل کند.
مراجع
[1] W.J. Boesch, J.S. Slaney, Preventing sigma phase embrittlement in nickel base superal- loys, Met. Progr. 86 (1964) 109–111.
[2] C.S. Barrett, Some industrial alloying practice and its basis, J. Inst. Met. 100 (1972) 65–73.
[3] For example, http://www.jst.go.jp/crds/pdf/2015/FR/CRDS-FY2015-FR-05.pdf.
[4] W. Hume-Rothery, G.V. Raynor, Structure of Metals and Alloys, fourth ed., Institute of Metals, London, 1962.
[5] L. Darken, R.W. Gurry, Physical Chemisry of Metals, McGraw-Hill, New York, 1953.
[6] H. Adachi, M. Tsukada, C. Satoko, Discrete variational Xα calculations. 1. Application to metal clusters, J. Phys. Soc. Jpn. 45 (1978) 875–883.
[7] H. Adachi, Introduction to Quantum Materials Chemistry—Approach from DV-Xα Method, Sankyo Publishing Co. Ltd., Tokyo, 1991 (in Japanese).
[8] M. Morinaga, N. Yukawa, H. Adachi, Alloying effect on the electronic structure of Ni3Al(γ0), J. Phys. Soc. Jpn. 53 (1984) 653–663.
[9] M. Morinaga, N. Yukawa, H. Ezaki, H. Adachi, New PHACOMP and Its Applications to Alloy Design, Superalloys 1984, Ed. by M. Gell et al., The Metallurgical Society of AIME, Warrendale, Pennsylvania, 1984, pp. 523–532.
[10] M. Morinaga, Alloy design based on molecular orbital method, Mater. Trans. 57 (3) (2016) 213–226.
[11] R.S. Mulliken, Electronic population analysis on LCAO-MO molecular wave functions. 1.,2.,3.,4, J. Chem. Phys. 23 (1955) 1833–1840. pp. 1841–1846, 2338-2342, 2343–2346.
[12] M. Morinaga, Y. Murata, H. Yukawa, Recent Progress in the New PHACOMP Approach, Materials Design Approaches and Experiences, ed. by J.-C. Zhao, M. Fahrmann and T. M. Pollock, TMS, Warrendale, Pennsylvania, 2001, pp. 15–27.
[13] M. Morinaga, N. Yukawa, H. Ezaki, H. Adachi, Solid solubilities in transition-metal-based
f.C.C. alloys, Philos. Mag. A 51 (1985) 223–246.
[14] M. Morinaga, H. Ezaki, H. Adachi, Solid solubilities in nickel-based f.C.C. alloys, Philos. Mag. A 51 (1985) 247–252.
[15] H. Ezaki, M. Morinaga, N. Yukawa, H. Adachi, Prediction of the occurrence of the σ phase in Fe-Cr-Ni alloys, Philos. Mag. A 53 (1986) 709–716.
[16] H. Ezaki, M. Morinaga, N. Yukawa, H. Adachi, Solid solubilities of molybdenum and tungsten in FCC transition-metal alloys, Philos. Mag. A 65 (1992) 1249–1260.
[17] P. Caron, T. Khan, Design of Superalloys for Single Crystal Blade Applications: A 20-Year Experience, Materials Design Approaches and Experiences, ed. by J.-C. Zhao, M. Fahrmann and T. M. Pollock, TMS, Warrendale, Pennsylvania, 2001, pp. 1–14.
[18] N. Yukawa, M. Morinaga, Y. Murata, H. Ezaki, S. Inoue, in: D.N. Duhl et al., (Ed.), High Performance Single Crystal Superalloys Developed by the d-Electrons Concept, Superal- loys, 1988 The Metallurgical Society of AIME, Warrendale, Pennsylvania, 1988, pp. 225–234.
[19] K. Matsugi, Y. Murata, M. Morinaga, N. Yukawa, Realistic Advancement for Nickel Based Single Crystal Superalloys by the d-Electrons Concept, Superalloys 1992, Ed. By S. D. Antolovich et al., TMS, Warrendale, Pennsylvania, 1992, pp. 307–316.
[20] R. Hashizume, A. Yoshinari, T. Kiyono, Y. Murata, M. Morinaga, K.A. Green et al., (Ed.), Development of Ni-Based Single Crystal Superalloys for Power-Generation Gas Turbines, Superalloys 2004, TMS, Warrendale, Pennsylvania, 2004, pp. 53–62.
[21] M. Morinaga, N. Yukawa, H. Adachi, Electronic structure and phase stability of titanium alloys, Tetsu Hagane 72 (1986) 555–562 (in Japanese).
[22] M. Morinaga, N. Yukawa, T. Maya, K. Sone, H. Adachi, in: Theoretical design of titanium alloys, Proc. Sixth World Conf. on Titanium, Cannes, France, June 6–9, 1988, Soci´et´e Franc¸aise de M´etallurgie, Paris, 1988, pp. 1601–1606.
[23] H. Okamoto, Phase Diagrams for Binary Alloys, second ed., ASM International Metals Park, OH, 2010.
[24] M. Morinaga, M. Kato, T. Kimura, M. Fukumoto, I. Harada, K. Kubo, in: Theoretical design of β-type titanium alloys, Proc. 7th World Conference on Titanium, San Diego, California, June 29–July 2, 1992, TMS, Warrendale, Pennsylvania, 1992, pp. 217–224.
[25] M. Morishita, Y. Ashida, M. Chikuda, M. Morinaga, N. Yukawa, H. Adachi, Active cor- rosion rate for Ti-based alloys in aqueous corrosion and its correlation with the bond order obtained by electron theory, ISIJ Int. 31 (1991) 890–896.
[26] M. Morishita, M. Chikuda, Y. Ashida, M. Morinaga, N. Yukawa, H. Adachi, Electronic states of the cathodes of titanium-based alloys in aqueous corrosion, Mater. Trans. JIM 32 (1991) 264–271.
[27] Y. Okazaki, A. Ito, T. Tateishi, Y. Ito, Effect of alloying elements on anodic polarization properties of titanium alloys in acidic solutions, Mater. Trans. JIM 35 (1) (1994) 58–66.
[28] M. Semlitsch, F. Stab, H. Webber, Titanium-aluminium-niobium alloy, development for biocompatible, high strength surgical implants, Biomed. Tech. 30 (1985) 334–339.
[29] D. Kuroda, M. Niinomi, M. Morinaga, Y. Kato, T. Yashiro, Design and mechanical properties of new β type titanium alloys for implant materials, Mater. Sci. Eng. A 243 (1998) 244–249.
[30] T. Saito, T. Furuta, J.H. Hwang, S. Kuramoto, K. Nishino, N. Suzuki, et al., Multifunctional alloys obtained via a dislocation-free plastic deformation mechanism, Science 300 (2003) 464–467.
[31] M. Abdel-Hady, K. Hinoshita, M. Morinaga, General approach to phase stability and elastic properties of β-type Ti-alloys using electronic parameters, Scr. Mater. 55 (2006) 477–480.
[32] H. Ikehata, N. Nagasako, T. Furura, A. Fukumoto, K. Miwa, T. Saito, First-principles cal- culations for development of low elastic modulus Ti alloys, Phys. Rev. B 70 (2004) 174113.
[33] L.A. Matlakhova, A.N. Matlakhova, S.N. Monteiro, S.G. Fedotov, B.A. Goncharenko, Properties and structural characteristics of Ti-Nb-al alloys, Mater. Sci. Eng. A 393 (2005) 320–326.
[34] Y.L. Zhou, M. Niinomi, T. Akahori, Effects of ta content on Young’s modulus and tensile properties of binary Ti-ta alloys for biomedical applications, Mater. Sci. Eng. A 371 (2004) 283–290.
[35] T. Ozaki, H. Matsumoto, S. Watanabe, S. Hanada, Beta Ti alloys with low young’s mod- ulus, Mater. Trans. 45 (8) (2004) 2776–2779.
[36] J.I. Kim, H.Y. Kim, H. Hosoda, S. Miyazaki, Shape memory behavior of Ti-22Nb- (0.5–2.0)O (at%) biomedical alloys, Mater. Trans. 46 (4) (2005) 852–857.
[37] J.I. Qazi, B. Marquardt, H.J. Rack, High-strength metastable beta-titanium alloys for bio- medical applications, JOM 56 (11) (2004) 49–51.
[38] S. Ishiyama, S. Hanada, O. Izumi, Effect of Zr, Sn and al additions of deformation mode and Beta phase-stability of metastable beta Ti alloys, ISIJ Int. 31 (1991) 807–813.
[39] N. Sakaguchi, M. Niinomi, T. Akahori, T. Saito, T.Furuta, Effects of alloying elements on elastic modulus of Ti-Nb-ta-Zr system alloy for biomedical applications, Mater. Sci. Forum 449–4 (2004) 1269–1272.
[40] N. Sakaguchi, M. Niinomi, T. Akahori, Tensile deformation behavior of Ti-Nb-ta-Zr bio- medical alloys, Mater. Trans. 45 (4) (2004) 1113–1119.
[41] M. Tahara, H.Y. Kim, T. Imamura, H. Hosoda, S. Miyazaki, Lattice modulation and super- elaticity in oxgen-added beta-Ti alloys, Acta Mater. 59 (2011) 6208–6218.
[42] J.I. Kim, H.Y. Kim, T. Inamura, H. Hosoda, S. Miyazaki, Shape memory characteristics of Ti-22Nb-(2–8)Zr(at%) biomedical alloys, Mater. Sci. Eng. A 403 (2005) 334–339.
[43] H.Y. Kim, H. Satoru, J.I. Kim, H. Hosoda, S. Mitazaki, Mechanical properties and shape memory behavior of Ti-Nb alloys, Mater. Trans. 45 (7) (2004) 2443–2448.
[44] Y. Fukui, T. Inamura, H. Hosoda, K. Walashima, S. Miyazaki, Mechanical properties of a Ti-Nb-Al shape memory alloy, Mater. Trans. 45 (4) (2004) 1077–1082.
[45] M. Morinaga, Y. Murata, H. Yukawa, Molecular orbital approach to alloy design, Applied Computational Materials Modeling-Theory, Simulation and Experiment, ed. by G. Bozzolo et al., Springer, New York, 2007, pp. 255–306.